
ДРУГИ СЕМИНАРСКИ РАБОТИ
- МЕДИЦИНА: |
||||||||||||||||||||
|
||||||||||||||||||||

НЕМЕНДЕЛИЗAМ
Генетиката e наука која ги проучува наследувањето и менливоста на особините кај живите организми. Таа проучува како биолошките карактеристики се пренесуваат од родителите на потомството. Наслелдувањето во најопшт смисол е процесс на предавање на својствата (генетска информација) во индивидуалниот развиток на потомствата (од генерација на генерација). Менливоста односно варијабилноста на својствата ги подразбира разликите кои постојат меѓу родителите и потомците. Варијабилноста на својствата, односно разликите кои постојат генетиката настојува да ги пронајде причините односно механизмите за настанување на варијабилност. Родителските својства кои се среќаваат кај потомците претставуваат единствено испреплетување на наследните фактори кои во многу зависат од факторите на средината. Според тоа човекот кој е предмет на хуманата генетика е продукт на меѓуиграта на наследување и средината. Предмет на проучување на генетиката е наследноста, почнувајки од мнолекуларната природа на генетскиот материал, начинот на кој гените ја контролираат функцијата на организмот па се до дистрибуција и однесување на гените во популациитге на организмите. Многу научни работници ја проучувале генетиката, меѓу кои е и Gregor Mendel кој ги поставил основите на теоријата на партикуларното наследување, односно концепција дека карактеристиките на организмите се одредени од наследните фактори или гени кои се пренесуваат низ генерациите без меѓусебно мешање. Науката која го проучувала наслелдувањето била наречена менделизам. Покрај менделовото наследување постои наследување кое се вика неменделово наслелдување или неменделизам.НЕМЕНДЕЛИЗЛМ
Многу голем број на заболувањса не ги следат класичните шеми на Менделовото наследување. Откриени се повеќе нови механизми: Полигенско мултифакториелно наслелдување, антиципација, геномски импринтинг, митохондријално наследување, спорадично наследување, мозаицизам и други.
ПОЛИГЕНСКО-МУЛТИФАКТОРИЕЛНО НАСЛЕДУВАЊЕТоа е таков вид на наследување каде што за изразеност на одредено својство потребно е взаемно дејство на два или повеќе гени и влијание на надворешни чинители: темперамент, интелигенција а наслелдувањето на болестите е поврзано со дејство на повеќе мутирани патолошки гени на различни локуси од кои секој има релативно мал ефект. Генетската компонента е полигенска - ендогена.
Всушност конституционална со карактер на кумулирање на ефектите. Во комбинација компонентите пробиваат одреден праг и кај човекот болеста се изразува фенотипски. Таа почесто се јавува во една фамилија, отколку во општата популација.
Изразувањето на болестите или својствата по закон на случајноста ќе се остварат со комбинација на поголем број гени кои имаат мал поединечен ефект. Такви гени се наоѓаат на различни генски локуси на хромозомите. За да може да дојде до нивна изразеност потребно е меѓусебно да се комбинираат. Таква меѓусебна комбинација би создала предиспозиција за хередитарна болест, односно доаѓа до експресија на болеста после раѓањето. Предиспозициите се промени во геномот кои немаат фенотипски ефект, но кои можат да се пренесат во потомството, а за да дојде до експресија потребно е присуство на надворешни фактори. Ризикот за повторно јавување во една фамилија е 3%.
Мултифакториелните болести почесто се јавуваат во една фамилија отколку во општата популација и ги карактеризира етиолошка хеторогеност.
Овие болести наследени од овој тип трансмисија не ги следат Менделовите закони на наследување.
Л тоа се: расцеп на уста, вродено исчашување на колковите, хипертрофична стеноза на пилорусот, улкусна болест, хипертензија, атеросклероза, алергиски болести, автоимуни заболувања, манична депресија, сенилна деменција, мултипска склероза, тумори и
другоСпорадично наследување
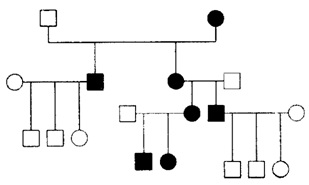
Заболувања кои неочекувано се јавуваат и кои во генетиката претставуваат
специфичен проблем се спорадични облици на наслелдување (сл. 1.1).
Не секогаш со вршење на анализа на историјата на болеста и бременостите во рамките
на родословот како и анализите на релевантните навидум својства кај останатите
членови на фамилијата, може да дојде до разоткривање на етиолошката поврзаност со
определени средински фактори. Или пак да се посочи на фамилијарна
Особеност на заболувањето.
Таквите етиолошки спорадични наслелдувања може да се последица на нови мутации, автосомно рецисивни наследувања, унипарентални дисомии, герминативен мозаицизам и тератогени ефекти
Сл. 1.1. Спорадично наследување
МИТОХОНДРИЈАЛНО НАСЛЕДУВАЊЕЕдинствен хромозом кој се наоѓа во цитоплазмата е митохондријален хромозом т.н. "25 хромозом" со добро позната генска мапа. Митохондријалната DNA (mt DNA) има 16.000 нуклеотидни парови со познат редослед и е во форма на прстен. Митохондријалниот хромозом Mt DNA учествува со 0,3% во целокупната клеточна DNA. Во mt DNA сите гени се добро дефинирани и имаат одговорност за кодирање на 22 транспарентни RNA кои се неопходни за митохондријална протеинска синтеза, две рибозомални RNA (12S и 16S) и 13 протеини кои се одговорни за оксидативна фосфорилација.
Митохондријалното наследување е од мајчинско потекло затоа што јајце клетката при оплодувањето останува целосна, задржувајки ја цитоплазмата, додека сперматозоидот при оплодувањето ја губи цитоплазмата. Затоа митохондријалното наследување се вика и мајчинско или цитоплазмично наслелдување.
Митохондријалното наслелдување mt.DNA се карактеризира со еднаква застапеност на двата пола, кои се зафатени подеднакво често и се со ист интензитет, така што и кај женските потомци може да дојде до заболување. Машките индивидуи не можат да ја пренесат болеста.
Варијабилноста на фенотипот е чиста. Зафатени со мутација на 25-от хромозон различните ткива се манифестираат со абнормален фенотип.
Митохондријалните мутации се одразуваат прво на клетките и ткивата со високи енергетски потреби, пред се на мозокот и мускулите, кај што се одвиваат интензивни митохондријални и енергетски оксидативни функции.
Митохондријалните мутациии можат да доведат до состојба на "хетероплазма" кај што има повеќе оде ден вид на митохондријални DNA молекули во клетка. "Хомоплазмични" клетки се оние во кои сите митохондријални имаат еднаква DNA. Хомоплазматичната DNA и хетероплазмичната DNA се разликуваат во различни ткива и може да се промени со возраста со компликација на клиничката слика.Честопати први ги покажуваат клиничките промени мускулното и нервното ткиво.Мутациите на митохондријалната DNA се случува 10 пати почесто од тие во нуклеарните гени кои се вклучени во оксидативна фосфилација. За ваквата појава причина може да биде намалената веродостојност на репликацијата и недоволно ефикасната репарација на митохондријалната DNA.
Пред 15 години се откриени митохондријалните заболувања и се во фокусот на интерес на генетичарите и лекарите. Досега се идентификувани 59 вакви заболувања а прва откриена митохондријална болест била Leber-овата оптичка атрофија во 1988 година. Митохондријалните заболувања може да се класифицираат во неколку групи и тоа: Во првата група припаѓаат оние болести што се должат на специфични мутации на митохондријалната DNA.MERRF синдромот (myoclonic epilessy and ragged red fibers), MELAS синдромот (myopathy, encephalomyopathy, Lactic acidosis, stroke), NARP синдромот (neurogenic muscule weakness, ataxia, and retinitis pigmentosa), Leber -овата оптичка невропатиа е мала група на случаи со Leigh-ов синдромот наследени од мајката.
Горенаведените болести се наследуваат од мајката и обично се должат на делеции или дупликации на митохондријалната DNA, предизвикувајки миопатии со или без офталмоплегија каде што припаѓа Kearns-овиот синдром.
Во втората група на митохондријални заболувања генетскиот дефект се среќава во јадрените гени што ги кодираат субединиците, на митохондријалната DNA. Во оваа група спаѓаат некои случаи на Leigh-ов синдром, Alpers-ови полидрофии, мио¬неврогастро-интестиналниот синдром, Friedreich-овата атаксија и Barth-овиот синдром. Генетичарите укажуваат дека подгрупи на некои заболувања се причинети од мутации на митохондријалната DNA, како што се глувост, наследни кардиомиопатии и diabetes mellitus a некои дефекти во митохондријалната DNA сугерираат промени во исходот на болеста кои се примарна причина од други фактори се вбројуваат Alzheimer-овата и Parkinson-овата болест. (сл.1.2),
Сл. 1.2. Митохондријално наследување
АНТИЦИПАЦИЈА
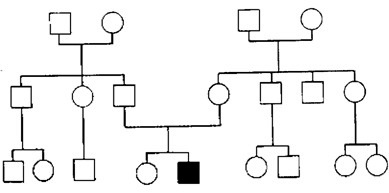
Кај некои автосомно доминантни заболувања како што се миотонична дистрофија (МД) и Huntington-овата болест, појавата на болест порано се појавува кај децата отколку кај нивните родители или болеста е со потешка клиниа слика во слелдните генерации. Таквиот феномен е наречен антиципација (сл.1.3).
Се смета дека овој ефект е резултат на предрасудите во утврдувањето кое се должи на начинот на селекција на семејствата. Додека пак некои истражуваи укажуваат дека тоа е артефакт на подобра опсервација и клиничка дијагноза во поново време. Заболувањето кое порано било дијагностицирано на 60 години сега поради подобри услови (подобри дијагностички апарати) ќе биде на 40 години. Набљудувањата на некои генерации укажуваат дека многу луѓе кои момнентално не биле заболени подоцна во животот кај нив се развила болеста. Кај индивидуи кај кои болеста се развила порано или била поизразена, многу полесно се откривала и овие индивидуи имале поголема можност и тенденција болеста да ја пренесат на нивните потомци.
Кај манично - депресивната психоза, шизофренијата и некои други е приметено ова налседување.
Лнтиципацијата има биолошка основа и е вистински биолошки феномен кој се јавува како резултат на експанзија на нестабилни повторувања на триплетни секвенции. За Huntington-овата болест се знае дека зависи од бројот на повторувања на тринуклеотидите ЦЛГ(цистонски аденински гванински) нуклеотид во молекулот на DNA во еден ген на хромозомот 4. Бројот на повторувањата е нестабилен и може да има зголемувања во идните генерации. Нормално луѓето имаат помеѓу 11 и 34 копии ЦЛГ триплета. Лко се најдат 39 повторувања во тие гени индивидуата ќе заболи од Huntington-ова болест на 75 години од својот живот, а додека во следните генерации овој број најверојатно ќе биде 40 при што ќе услови појава на болест на 59 години, а со повторување на болеста заболувањето ќе се јавува на 54 години и т.н. (2) Експанзијата на повторувањата на ЦТГ триплетот на 3' нетранслатирачкиот крај на генот за миотонична дистрофија се случува предоминантно во мајчината мејоза, така да со оваа појава се овозможува објаснување на тешките неонатални форми на миотичната дистрофија кога генот е наслелден од мајката. Неафектираните лица имаат 5 до 30 повторувања. Тие со 50-100 повторувања може да бидат умерено засегнати или да немаат симптоми. Оние со над 100 повторувања имаат класична слика на миотона дистрофија. Кај следните генерации бројот на повторување е се поголем. Кај умерено зафатен родител со 80 повторувања може да има потомци со преку 1000 повторувања. Слична експанзија на ЦЛГ триплетот на 5' крајот на генот за Huntington-овата болест го зголемува ризикот за појава на болест кај децата кога гените се наследени од татковата мејоза.
Сл. 1.3. Aнтиципација
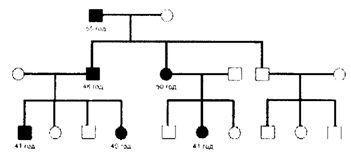
МОЗАИЦИЗАМВо организмот одредено ткиво може да содржи една или повеќе видови на клеточни линии поради грешка за време на митозата во било која фаза после концепцијата претставува мозаицизам. Зависно од клетките кои ги опфаќа се дели на соматски и герминативен и се смета за невообичаен начин на наследување на фенотипските карактеристики.
ГОНАДЕН МОЗАИЦИЗАМЛко генетските тестови се нормални и фенотипски родителите нормални а имаат повеќе од едно дете заболено таквата појава се објаснува со гонадниот мозаицизам. Такви појави се присутни при автосомно доминантните заболувања (ахондроплазија и osteogenesis imperfekta) I Х-врзано рецисивно нарушување (ducenne-ова мускулна дистрофија и хемофилија). Тие се објаснуваат со гонаден или герминален мозаицизам на еден од родителите. (настанува мутација во пропорција на половите клетки). Како добар пример за ова е мутација на генот за колаген одговорен за osteogenezis imperfekta во пропорција на сперматозоиди на клинички нормален татко кој имал две заболени деца со две различни партнерки.
Германитивен мозицизам е важен и треба да се има предвид при сметање на ризикот за повторното појавување на болеста во хенстското советување на новите автосомни доминантни (ЛД) И Х-рецисивно врзани ХР мутации.СОМАТСКИ МОЗАИЦИЗАМ
Генетската конституција на организмот која што поседува станици со различен генотип (Hall 1988) се нарекува соматски мозаицизам. Таа појава настанува со повеќе кратна мутација после концепција.Хромозомски мозаицизам не е редок посебно со гоносомопаптија. Мозаицизмот понекогаш може и да се изгуби и тоа обично кога клетката го започнува својот живот како анеуплоидна, а покасно со делбата на една од станиците на женската клетка се губи гоносом Х тој се нормализира а воедно го потиснува растот на другите анеуплоидни станици. Кај мозаицизмот на примерите 45, Х/47 ХХХ фенотипот на Турнерровиот синдром ќе биде поблаг, кај мозаичниот облик на Downov синдром клиничката слика ќе биде поблага доколку е поголем процентот на станицата без трисомија 21.
Посебно занимлив е соматскиот генски мозаицизам кога со мутација опфаќа повеќе гени кои се близу еден до друг. Генска соматска мутација множе да се случи секогаш после оплодување со тоа што може и да се промени генотипот и на аутосомот а воедно и на геносомот.
Последицата од мутацијата може да биде голема делеција на генот или некоја друга структурна генска промена. Понекогаш се открива склоност на поедини локуси наспроти соматската мутација, можно фенотипско влијание на соматска генска мутација зависи од времето на развојот на киематонезата и кога е дошло до мутација, ако е настаната генската мутација непосредно после оплодувањето, фенотипот ќе биде ист како генот наследен од родителите.
Лко мутираниот ген е сместен само во ткивото на постелката нема да има послелдици за фенотипот на плодот. Во патологијата на болеста кај човекот кои понекогаш зафаќаат одреден дел од телото на човекот се вбројуваат во послелдици од соматска генска мутација: хемихипертрофија, сегментна неурофиброматоза наведени како примери. Соматската мутација може да се пренесе на потомството ако има присуство на мозаицизам и во станиците на герминативниот епител, а тоа значи и во оогоните и сперматогониите (Hall 1988). (2)
Многу малигни заболувања во патологијата на индивидуате се последица на соматските хромозомски и генски мутации. Соматската мутасција може да биде пренесена на потомството со потполна експресија зависно од тоа дали мутацијата е присутна во сите или само во некои полови клетки.УНИПАРЕНТНА ДИСОМИЈА
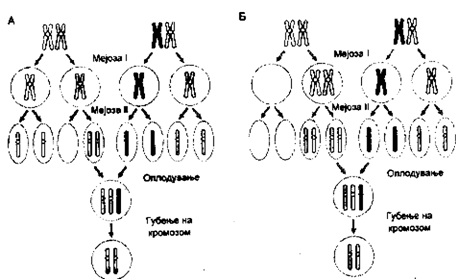
Секоја клетка со зрели гамете е дисомична, нив ги сочинуваат 23 хромозомски пара, така да еден хромозом во пар од мајката и другиот од таткото. Секоја индивидуал наследува еден пар од секој родител. Доколку хромозомите од парот потекнуваат од еден родител, мајката или таткото што се вика унипарентна дисомија бројот на хромозомите останува ист, а хромозомскиот пар е само од еден родител, или од мајката или само од таткото.
Изидосомија претставува посебен облик на унипарентна дисомија. Лко се случи една индивидуал да наследи две копии од ист хомолог, било да е од мајката или од таткото поради грешката во мејозата II тогаш тоа се нарекува унипарентна изидосомија. Но доколку индивидуате наследи два различни хомолога од едниот родител или мајката или таткото поради грешки во мејозата I тогаш за таа појава велиме дека се вика уиппарентна хетеро дисокмија. И во двата случаи зиготот ќе биде трисомичен и со губење на еден хромозом станува дисомичен.Aко една третина од овие губења на хромозомите се случат со еднаква фреквенција тогаш настанува унипарентна дисомија.
Постои и друго размислување од страна на генетичарите и тоа дека унипарентна дисомија може да настане како резултат на гаметата од едниот родител, која не содржи дел од хомологните хромозоми кое е нулисомично и која е фертилизирана со гамета која поради грешка во мејозата станала дисомична (сл.1.4).
Сл. 1.4 Унипарентна изидосомија - Унипарентна хетеродисомијаНајчесто унипарентна дисомија настанува со губење на еден хромозом од трисомичен зигот. Послелдицата ќе се разликува зависно од времето на настанување на оргиналната грешка или нераздлвојувањето, Зависно од тоа дали грешката настанала во мејоза I или во мејоза II (6)
Со користење на DNA технологија унипарентната дисомија се покажала како причина на таткото заболен од хемофилија кој имал два сина од кои едниот син исто заболен од хемофилија а другиот со цистична фиброза кое било родено од родители каде што мајката била носител (со докажано татковство).
ГЕНОМСКИ ИМПРИНТИНГГеномски импринтинг (Genomik imprinting) има поголемо значење во биологијата и генетиката. Тоа е новооткриена појава во која алелните гени кај цицачите се однесуваат различно зависно од тоа дали се работи за мајчините или таткови наследувања. Тој епигенетски процес се мисли дека има влијание на фенотипската експресија на некои генетски заболувања, зависно од тоа дали некоја болест е наследена од мајката или таткото.
За некои генски локуси многу е важно од кој родител потекнуваат, бидејки од тоа зависи јачината на фенотипската експресија. Механизмот на импринтингот се поврзува со метилацијата на DNA и некои други недоволно објаснети процеси кои веројатно се споредуваат со генската експресија. (Holliday 1989) Се мисли дека имипринтинг го зафаќа и ембрионот и постелката истовремено. Дефектот во тој процесс може да има големо јако дифузно влијание на растото и развојот. Мајчиниот и татковиот удел во некои делови на геномот се важни а по некогаш и комплементарни. Некои добро проучени синдроми во човековата патологија со различна фенотипска експресија можат да се објаснат со импринтинг. Се претпоставува дека импринтингот во гените на оварија има одредено влијание на развојот кај машкиот плод само тогаш ако е сместен во плодовиот тестис. Потомството на мајката заболено од миотона дистрофија во 50 % од случаите ќе биде зафатено со болест и тоа потежок со порано губење на фенотипот. Исто така се однесува и на генот на NF 1, наследен од мајка ген кој дава многу потешка клиничка слика. Обратно од ова Hungtingtonova болест е потешка, се појавата порано во потомства ако мутираниот ген кој потекнува од таткото.
Како добар пример се синдромите Angelman I Prader - Wili. И двата синдрома се предизвикани да со слична хромосомска делеција 15 q 11 -13 а се чини дека во игра се два различни гена на 15 q хромозоми.
Дилетирана регија на хромозоми многу е голема во молекуларна смисла со сигурно многу генски локуси. Меѓутоа ако делеција е од мајчино потекло, се развива фенотипот Angelman sindrom со делеција 15q. 11-13 на татковиот хромозом настанува фенотип Prader - Wily.Пoсмaтрajки го развојот на монофакторски и хромозомски абнормалности сигурно ќе се внесе нова светлина во објаснувањето на малигните болести. (Reik 1989) . И двете заболувања се сретнуваат приближно кај 1/15.000 индивидуи и двете заболувања околу 70% се предизвикани со хромозомски делеции (2) Молекуларната база на импринтингот сеуште останува нејасна. Од досега познати 16 гени кои се инпринтирани кај глушец и човек, се смета дека постои асоцијација меѓу метилацијата и транскипциската инактивација. Врзувањето на метилните групи за DNA може да го инхибира врзувањето на протеините што ја започнуваат транскрипцијата. Нема сознание дали метилацијата е примарен импринтинг сигнал или дали служи единствено да го подржува импринтинг сигналот кога тој е веќе воспоставен преку други механизми
ЗАКЛУЧОКОд горе напишаното можеме да заклучиме дека наследувањето е многу потребно за настанокот, растот и развојот на секоја единка. Меѓутоа луѓето се производ не само на генетските фактори туку и на околинските фактори, па затоа некои појави или болести се објаснуваат со неменделизам. Зборуваме за влијание од околината, зборуваме за мултифакторски условена сослтојба. За полигенско-мултифакторијално наследување можеме да кажеме дека е таков вид на наслелдување каде што за изразеност на одредено својстлво потгребни се взаемно дејство на две или повеќе гени и влијание на надворешни чинители, наследувањето е поврзано со дејствување на повеќе мутирани, патолошки гени на различни локуси од кои секој има релативно мал ефект. Болеста почесто се јавува во една фамилија отколку во општата популација. Спорадично наследување неочекувано се јавува. Со анализа на историја на болеста ирелевантникте својства на семејството може да се разоткрие етиолошка поврзаност со определени средимски фактори, а може да е послелдица на нови мутации. Наследувањето врзано за mt DNA е од мајчинско потекло, се карактеризира со подеднаква застапеност на двата пола така што и женските потомци можат да заболат, а машките индивидуи неможат да ја пренесат болеста на потомците. Различните ткива зафатени со мутација на 25 хромозом се манифестираат со абнормален фенотип. Лнтиципацијата има биолошка основа а се случува заради експанзија на нестабилни повторувања на триплетни секвенци. Мозаицизмот е наслелдување кое настанува како резултат наг решките во митоза во која било фаза при што одредено ткиво е составено од една или повеќе клеточни линии. Секој човек нормално наследува еден пар хомологни хромозоми од секој родител но ако не се случи да ги имаат наследено двата хомологни хромозоми оде ден пар само од едниот родител тогаш станува збор за унипарентна дисомија, ако се случи да наследи две копии на ист хомолог од еден родител заради грешка во мејоза-II тогаш станува збор за унипарентна изидисомија, ако пак наследи два различни хомолога од еден родител заради грешка во мејоза I тогаш станува збор за унипарентна хетеродисомија, во двата случаи зиготот ќе биде трисомичен и со губење на еден хромозом станува дисомичен. Карактеристично за геномски импринтинг е тоа што можат да се јават различни клинички карактеристики зависно од тоа дали генот е наслелден од таткото или мајката. Делеција од 3 до 4 mb на долниот крак на 15-от хромозом се наследи од таткото потомството манифестира Prader-Willi синдром, а кога делецијата е наследена од мајката потомството манифестира Angelman синдром. Болестите се предизвикани со хромозомски микро делеции.
КОРИСТЕНА ЛИТЕРАТУРА
1. Трајковски В. Хумана генетика. Филозовски факултет, Институт за дефектологија, Скопје, 2005; 136-142
2. Zargollern Lj I sur. Humana genetika. Medicinska naklada, Zagreb,1994; 35 38
3. Tucic N. Matic G.O genima I ljudima. Centar za primenjenu psihologiju, Beograd, 2002 ; 15-21
4. http:// www. people. virginia.edu
5. http:// www. ich. ucl. ac.uk
6. http:// www.lpch. orgDownload СЕМИНАРСКА РАБОТА у wordu » » »

Besplatni Seminarski Radovi - Бесплатно семинарска работа